

{kind=link}

Research presentation
Håkan Hugosson
Senior lecturer, docent
Research subject: Physics
Email: hakan.hugosson@hig.se
Telephone: +46 26 64 82 78
CURRENT RESEARCH
READ MORE ABOUT
Multi-Scale Dynamical Quantum Mechanical Simulations of Materials
The investigation of our physical surroundings by making quantum mechanical (QM) electronic structure calculations is a continually growing field of research. Electronic structure calculations have shown usefulness in many sciences, ranging from solid-state physics to pharmacology. They have not only increased understanding of the experimental results but have also shown the capability to predict key properties and therefore allow rational design of molecules and materials based on theoretical predictions. Electronic structure calculations have even breached into biology, the science of life itself. With the possible increased scientific understanding and applicability to problems in e.g. medicine and nanotechnology, electronic structure calculations carry enormous potential impact, also on the every-day world around us. The reach of electronic structure calculations is further enhanced in quantum or ab-initio molecular dynamics (QMD/AI-MD) where the full dynamics of the system at realistic temperatures are revealed.
One of the most important theoretical techniques in the study of large systems at room temperature, e.g. biological or polymer systems, is molecular dynamics (MD) simulations. In MD the system is described as a set of particles that are propagated according to the laws of classical mechanics over a given potential energy surface. Depending on the way in which the interaction potential between the particles is obtained, MD methods can be divided into classical force field MD or quantum MD methods. Though classical force field MD methods, the by-far most common approach, are very successful for the simulation of many biological and polymer systems, chemical reactions and more complex atoms such as transition metals can only be treated adequately by a complete quantum mechanical description. In QMD all the interactions are calculated from an electronic structure method (here density functional theory - DFT). QMD simulations are therefore parameter-free and enable a direct and potentially unbiased simulation of chemical and physical events. Since temperature is taken into account a sampling of the conformational space is made, making simulations less biased upon choices of initial conditions and chosen reaction coordinates.
Thermoelectric Polymers
To meet growing world-wide demand of electricity (projected to have doubled to 26TW by 2050) the search is on for alternative renewable energy sources convertible into electricity. Since the sun is the largest readily available source of energy, and one that easily could satisfy our energy needs with an irradiating power of the order of 100 000 TW, ways to convert solar energy into electricity are very attractive. Among the most promising such ways to convert solar heat into electricity is using thermoelectric generators (TEG). TEGs are constructed using thermoelectric materials, materials where a temperature gradient gives rise to an electric current. Among the drawbacks of the presently used inorganic thermoelectric materials are that they are composed of hazardous elements with low natural abundance. Organic polymer-based thermoelectric materials on the other hand, though presently being less efficient, can be mass-produced using safer highly abundant elements at low cost.
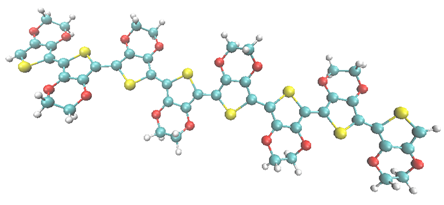
Figure 1. The 8-oligomer of the polymer poly (3,4-ethylenedioxythiophene) - PEDOT caught in the fly during Quantum MD simulations. Carbon atoms are shown in blue, oxygen atoms in red, sulphur atoms in yellow, and hydrogen atoms in white.
Thermoelectric devices consist of many thermoelectric couples (one material with n-type negative charge carriers and the other with p-type positive charge carriers) wired electrically in series and thermally in parallel. In order to attain a sizable Seebeck voltage a large temperature gradient needs to remain over time across the thermocouple. We therefore need both:
- a high Seebeck co-efficient (to generate a large Seebeck voltage)
- high electrical conductivity (to easily move the charge carriers)
- low thermal conductivity (to maintain the thermal gradient).
These three characteristics are not easily combined, e.g. metals are both highly conducting to heat and electricity, and thus good thermoelectric materials are a special class of materials. Among the organic polymer-based thermoelectric materials that are subject to intense interest in the scientific community, doped poly(3,4-ethylenedioxythiophene) (PEDOT), see figure 1, with thermoelectric power, tunable high electric conductivity, and air stability is widely used and has emerged as a most promising candidate.
Our simulations of PEDOT
Current strategies to increase its thermopower include doping with anions, e.g. polystyrene sulfonate (PSS), tosylate ions (Tos), or transition metal ions. In the present simulations we study both uncharged and charged, pristine and doped with anions, PEDOT oligomers, in the polymeric (1D chain), and in the 3D crystal state using quantum molecular dynamics. Despite the wide application of PEDOT and wealth of data from various experimental techniques, atomic scale descriptions of its electronic and geometric structures in general remain incomplete and especially the charge carrying polarons in these systems are still poorly understood.