

{kind=link}

Forskarpresentation
Håkan Hugosson
Universitetslektor, docent
Forskningsämne: Fysik
E-post: hakan.hugosson@hig.se
Telefon: 026-64 82 78
AKTUELL FORSKNING
Moderna elektronstrukturs-beräkningar tar fram elektronernas kvantmekaniska banor runt atomerna och är grunden för komplexa datorsimuleringar med vars hjälp man teoretiskt kan bestämma egenskaper hos material och molekyler. Här används denna sorts beräkningar, som görs på massivt parallella datorer vid nationella och internationella superdatorcenter, till att finna sätt att förbättra egenskaperna hos s.k. termoelektriska polymerer, en sorts material som omvandlar värmeskillnader till den elektricitet vi behöver för en omställning till mer hållbar energiframställning.
LÄS MER OM
Dynamiska kvantmekaniska simuleringar av material i flera skalor
Undersökningen av vår fysiska omgivning genom kvantmekaniska (QM) beräkningar av elektronstrukturerna i material och molekyler är ett ständigt växande forskningsområde. Elektronstrukturberäkningar har visat sig vara användbara inom många vetenskaper; från fasta tillståndets fysik till farmakologi. De har inte bara ökat förståelsen för de experimentella resultaten utan har också visat att de kan förutsäga viktiga egenskaper och därmed möjliggöra rationell utformning av molekyler och material på grundval av dessa teoretiska förutsägelser. Beräkningar av elektroniska strukturer har till och med etablerat sig i biologin, själva livets vetenskap. I och med denna ökade vetenskapliga förståelse och möjliga tillämpningar på utmaningar inom t.ex. medicin och nanoteknik har beräkningar av elektroniska strukturer en enorm potentiell inverkan, även på den vardagliga världen runt omkring oss.
Elektronstrukturberäkningars räckvidd ökar ytterligare i kvant- eller ab-initio-molekylär dynamik (QMD/AI-MD) där systemets hela dynamik, atomernas rörelser, vid realistiska temperaturer simuleras fram. En av de viktigaste teoretiska teknikerna för att studera stora system vid rumstemperatur, viktigt för t.ex. biologiska eller polymera system, är s.k. molekyldynamiska simuleringar (MD). I MD beskrivs systemet som en uppsättning partiklar som förflyttas enligt den klassiska mekanikens lagar över en given potentialenergiyta. Beroende på hur växelverkan mellan partiklarna erhålls kan MD-metoderna delas in i klassiska kraftfälts MD-metoder eller kvantmekaniska MD-metoder. Även om klassiska MD-metoder med kraftfält, som är den absolut mest använda metoden, är mycket framgångsrika för simulering av många biologiska och polymera system, kan kemiska reaktioner och mer komplexa atomer, t.ex. övergångsmetaller, endast behandlas på ett tillfredsställande sätt med hjälp av en fullständig kvantmekanisk beskrivning. I kvantmekaniska MD-metoder (QMD) beräknas alla interaktioner från en elektronstrukturmetod (här densitetsfunktionalteori - DFT). QMD-simuleringar är därför parameterfria och möjliggör en direkt och potentiellt opartisk simulering av kemiska och fysiska händelser. Eftersom systemens temperatur beaktas görs ett urval av konformationsrummet, möjliga geometrier, vilket även gör att simuleringarna inte påverkas lika mycket av val av till exempel utgångsförhållanden och valda reaktionskoordinater.
Termoelektriska polymerer
För att tillgodose den ökande efterfrågan på elektricitet i världen (som beräknas ha fördubblats till 26 TW år 2050) söker vi ständigt efter alternativa förnybara energikällor som kan omvandlas till el. Eftersom solen är den största tillgängliga energikällan, som lätt skulle kunna tillgodose våra energibehov med en strålningseffekt i storleksordningen 100 000 TW, är det mycket lockande att omvandla solenergi till elektricitet. Ett av de mest lovande sätten att omvandla solvärme till elektricitet är att använda termoelektriska generatorer (TEG). TEG:er konstrueras med hjälp av termoelektriska material, material där en temperaturskillnad ger upphov till en elektrisk ström i materialen den så kallade Seebeck-effekten. Nackdelarna med de oorganiska termoelektriska material som för närvarande används är att de består av giftiga grundämnen med låg naturlig förekomst. Organiska polymerbaserade termoelektriska material å sin sida är för närvarande visserligen mindre effektiva, men kan massproduceras med hjälp av säkrare, mycket rikliga grundämnen till en låg kostnad.
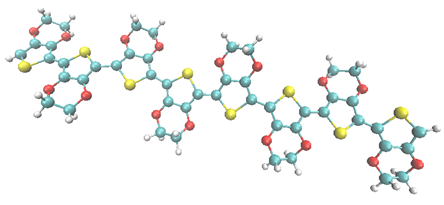
Figur 1: 8-oligomeren av polymeren poly (3,4-etylendioxytiofen) - PEDOT fångad i sin rörelse under kvantmekanisk MD-simuleringarna. Kolatomer visas i blått, syreatomer i rött, svavelatomer i gult och väteatomer i vitt.
Vi behöver därför material med både:
- en hög Seebeckkoefficient (för att generera en stor Seebeckspänning)
- hög elektrisk ledningsförmåga (för att lätt flytta laddningsbärarna)
- låg värmeledningsförmåga (för att bibehålla temperaturskillnaden).
De dessa tre egenskaper kombineras inte lätt; metaller är t.ex. både starkt värmeledande och goda elektriska ledare, och därför är bra termolelektriskt material svårfunna. Bland de organiska polymerbaserade termoelektriska material som är föremål för ett intensivt intresse i forskarsamhället används i stor utsträckning dopad poly (3,4-etylendioxytiofen) (PEDOT), se figur 1, som har termoelektrisk förmåga, hög elektrisk ledningsförmåga, låg temperaturledning och sönderfaller inte i luft. PEDOT framstår därför som en mycket lovande kandidat för att vara materialet i framtida termoelektriska generatorer.
Våra simuleringar av PEDOT
För närvarande omfattar strategierna för att öka PEDOT:s termiska effekt bl.a. dopning med anjoner, t.ex. polystyrensulfonat (PSS), tosylatjoner (Tos) eller övergångsmetalljoner. I våra simuleringar studerar vi både oladdad och laddad PEDOT, odopat och dopat med anjoner, samt PEDOT-oligomerer i polymeriskt tillstånd (1D-kedja) och i 3D kristalltillstånd med hjälp av kvantmekanisk molekylärdynamik. Trots omfattande användning av PEDOT och en mängd data från olika experimentella tekniker är beskrivningarna på atomär skala av dess elektroniska och geometriska strukturer fortfarande ofullständiga, och särskilt de laddningsbärande polaronerna i dessa system är inte fullständigt förstådda.